The Plesa Lab focuses on accelerating the pace at which we understand and engineer biological systems, particularly proteins. Towards this end, we develop new technologies for gene synthesis, multiplex functional assays, in-vivo mutagenesis, genotype-phenotype linkages, and computational approaches to explore high-dimensional datasets of sequence-function relationships. These allow us to both access the huge sequence diversity present in natural systems as well as carry out testing of rationally designed hypotheses encoded onto DNA at much larger scales than previously possible. Using these approaches, we can quickly characterize and engineer entire protein families, rather than focusing on individual proteins. Our work is highly-interdisciplinary and touches on aspects from many fields including biochemistry, synthetic biology, molecular biology, microbiology, structural biology, genetics, bioinformatics, and quantitative biology.
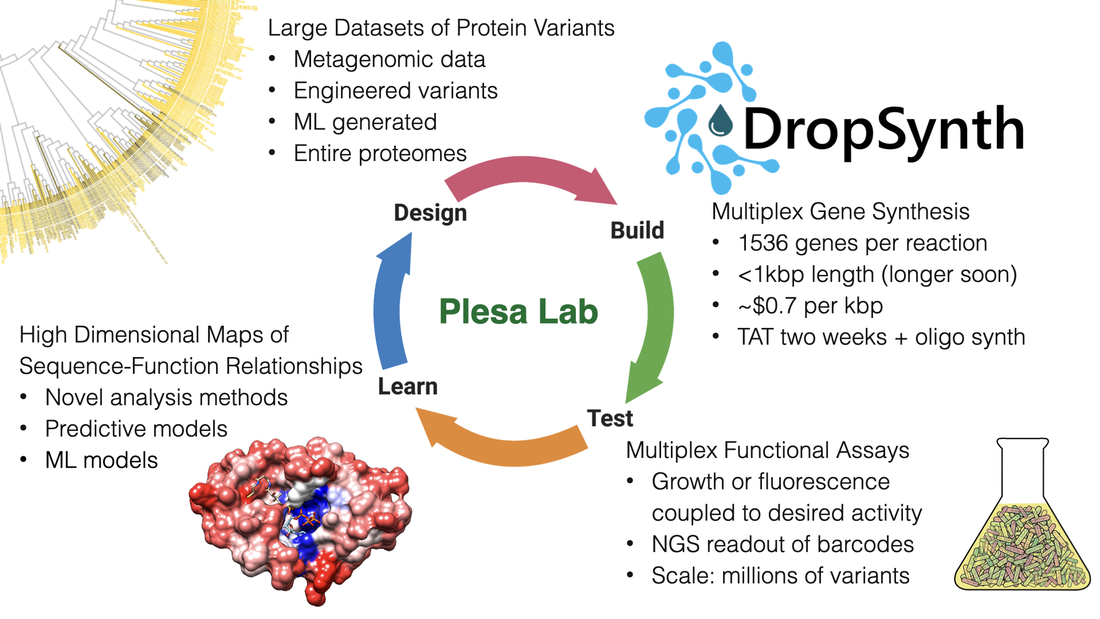
Join us as we delve into the mysteries of biology where traditional methods often fall short due to the complexity and diversity of natural systems. In our lab, we tackle these challenging inquiries by designing and synthesizing extensive libraries of protein coding genes using DropSynth multiplex gene synthesis, a low-cost scalable method which we are constantly improving.
As a part of our team, you'll have the opportunity to characterize these libraries using multiplexed in-vivo assays, coupling function to a simple sequencing-based readout. This enables us to derive fitness scores for each variant, helping us create predictive models of sequence-function relationships and expanding the forefront of synthetic biology.
|
Gene Synthesis
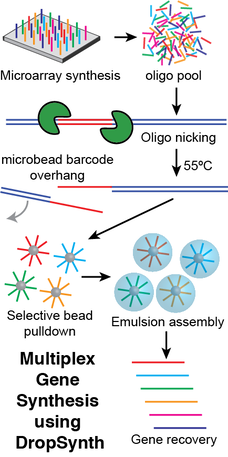
We are developing new technologies to synthesize large DNA libraries. Biological systems are incredibly complex. Successfully understanding and engineering these systems on reasonable timescales requires large-scale testing of DNA-encoded hypotheses. To this end, we have developed technologies, such as DropSynth, to synthesize libraries of thousands of genes at low cost. DropSynth uses bead-based barcode hybridization to pull down the microarray-derived oligonucleotides necessary for a gene’s assembly, which are then processed and assembled in water-in-oil emulsions. This approach was previously used to assemble libraries of over >7,000 synthetic genes at a cost below $2 per gene (Plesa et al., Science 2018). We are actively developing new gene synthesis technologies with the goals of:
|
|
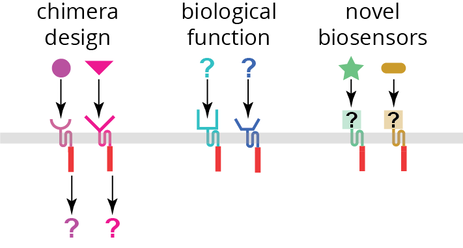
Chimeric Histidine Kinases for Biosensing
We are harnessing the immense natural diversity of receptor histidine kinases to build new biosensors. Bacteria have evolved a multitude of receptor histidine kinases to sense their outside world. These sensors, part of two-component systems, can detect many different small-molecule ligands and stimuli including nutrients, pH changes, quorum signaling molecules, metal ions, antibiotics, and other toxic compounds. Although hundreds of thousands of histidine kinases have been identified, with more being found regularly, very few of these have been characterized. Deciphering the function of members in this large protein family on a reasonable timescale will require the ability to experimentally characterize these systems at large scales. Our lab is building a platform to allow histidine kinase sensory domains to be characterized and engineered en masse by coupling them to a standardized output. This will allow us to both characterize their biological function and engineer novel receptors for ligands of interest against which natural sensors do not exist.
We are harnessing the immense natural diversity of receptor histidine kinases to build new biosensors. Bacteria have evolved a multitude of receptor histidine kinases to sense their outside world. These sensors, part of two-component systems, can detect many different small-molecule ligands and stimuli including nutrients, pH changes, quorum signaling molecules, metal ions, antibiotics, and other toxic compounds. Although hundreds of thousands of histidine kinases have been identified, with more being found regularly, very few of these have been characterized. Deciphering the function of members in this large protein family on a reasonable timescale will require the ability to experimentally characterize these systems at large scales. Our lab is building a platform to allow histidine kinase sensory domains to be characterized and engineered en masse by coupling them to a standardized output. This will allow us to both characterize their biological function and engineer novel receptors for ligands of interest against which natural sensors do not exist.
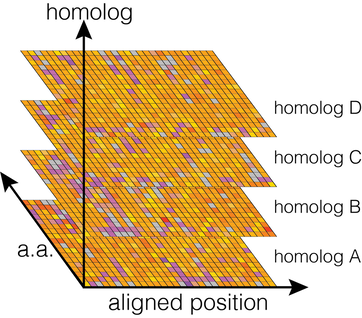
Broad Mutational Scanning
We are broadly interested in questions around the exploration of sequence space. Functional sequence space can be highly degenerate, containing many homologs with near identical functions despite having highly-divergent sequences as a result of evolutionary processes. Broad Mutational Scanning is a method to probe sequence-function relationships throughout an entire, functionally-related, protein family by assessing the impact of mutations around many different evolutionarily-related homologs simultaneously. We’re interested in addressing a number of open questions in this area:
We are broadly interested in questions around the exploration of sequence space. Functional sequence space can be highly degenerate, containing many homologs with near identical functions despite having highly-divergent sequences as a result of evolutionary processes. Broad Mutational Scanning is a method to probe sequence-function relationships throughout an entire, functionally-related, protein family by assessing the impact of mutations around many different evolutionarily-related homologs simultaneously. We’re interested in addressing a number of open questions in this area:
- How well do fitness landscapes of homologs correlate as a function of evolutionary divergence?
- How do we build predictive models of sequence-function relationships within protein families sharing function?
- How many functionally related sequences need to be experimentally characterized before we can accurately predict the function of a related but uncharacterized sequence or mutation? is accurate prediction even possible?
- Given a priori knowledge (sequence conservation, co-evolutionary couplings between residues, homology modeling, etc…), how do we focus our limited experimental measurements on the most informative areas?
Machine Learning Approaches for Exploring Sequence Space and Protein Engineering
Recently machine learning (ML) methods have made great progress in finding underlying patterns in sequence data to help guide limited experimental resources to the most promising sequences and even generate completely novel sequences distant from any input and potentially inaccessible by evolutionary processes. We are combining our unique gene synthesis capabilities and multiplexed functional assays to guide machine learning approaches and evaluate the key parameters affecting their performance.
Recently machine learning (ML) methods have made great progress in finding underlying patterns in sequence data to help guide limited experimental resources to the most promising sequences and even generate completely novel sequences distant from any input and potentially inaccessible by evolutionary processes. We are combining our unique gene synthesis capabilities and multiplexed functional assays to guide machine learning approaches and evaluate the key parameters affecting their performance.
Large-Scale Antibody Generation
We are developing methods to generate antibodies and antibody mimetics against thousands of antigens at once and carry out tests of cross-reactivity.
We are developing methods to generate antibodies and antibody mimetics against thousands of antigens at once and carry out tests of cross-reactivity.

Synthetic Metagenomics
The advent of next-generation sequencing has lead to an exponential increase in the amount of digital sequence information available in metagenomic databases. Synthetic metagenomics involves identifying sequences of interest from these databases, using gene synthesis to build the corresponding DNA molecules, and characterizing these en masse using functional assays. This approach leverages natural diversity to identify the optimal sequences for some desired functionality.
The advent of next-generation sequencing has lead to an exponential increase in the amount of digital sequence information available in metagenomic databases. Synthetic metagenomics involves identifying sequences of interest from these databases, using gene synthesis to build the corresponding DNA molecules, and characterizing these en masse using functional assays. This approach leverages natural diversity to identify the optimal sequences for some desired functionality.
- Developing novel multiplex functional assays.
- Building novel library-on-library approaches.
- Designing novel methods for maintaining phenotype-genotype linkages.
- What are the factors affecting interspecies protein compatibility?
Antibiotic Resistance
The emergence of antibiotic resistance has typically been studied in the context of genes from individual pathogenic organisms as opposed to a more community-wide approach, which would better reflect natural diversity of targets affected by broad-spectrum antibiotics. Understanding how resistance emerges given different evolutionary starting points will give us fundamental insights into its spread. We develop methods to tackle such fundamental questions at large scales by synthesizing large libraries of genes encoding antibiotic targets, introducing them into a model organism, and screening them (and their mutants) for resistance. Our current work focuses on dihydrofolate reductase (DHFR), a major antibiotic target in the folate acid biosynthesis pathway which is required for nucleic acid synthesis.
The emergence of antibiotic resistance has typically been studied in the context of genes from individual pathogenic organisms as opposed to a more community-wide approach, which would better reflect natural diversity of targets affected by broad-spectrum antibiotics. Understanding how resistance emerges given different evolutionary starting points will give us fundamental insights into its spread. We develop methods to tackle such fundamental questions at large scales by synthesizing large libraries of genes encoding antibiotic targets, introducing them into a model organism, and screening them (and their mutants) for resistance. Our current work focuses on dihydrofolate reductase (DHFR), a major antibiotic target in the folate acid biosynthesis pathway which is required for nucleic acid synthesis.
- Can we predict which sequences encoding an antibiotic target will be susceptible and which mutations in these sequences are likely to confer resistance?
- Is resistance more likely to emerge in one homolog over another?
- Developing high-throughput screens for narrow spectrum inhibitors.